Информационные материалы
Изучение наследственной предрасположенности к многофакторным заболеваниям крайне важно для их диагностики и терапии. Для оценки роли наследственных факторов в развитии многофакторного заболевания, как правило, используют анализ сцепления, анализ ассоциации или экспериментальные модели развития заболевания на животных [1].
Большую практическую ценность представляет исследование полиморфных маркеров в генах–кандидатах, продукты которых вовлечены в патогенез многофакторного заболевания [1].
Анализ сцепления основан на определении вероятности совместного наследования фенотипического признака (заболевания) и исследуемого маркера в семье. При этом исследуют совместную сегрегацию генов при передаче от родителей к потомкам в ряду поколений [1].
При использовании метода идентичных по происхождению аллелей (IBD, identical by descent) информацию о сцеплении получают в результате анализа наследования маркеров в парах больных родственников без предварительного выбора типа наследования и других характеристик. Данный подход заключается в оценке того, насколько чаще по сравнению со случайной сегрегацией пара больных родственников (чаще всего для анализа используют сибсовые пары) наследует один и тот же аллель. Анализ сцепления может проводиться с исследованием как ядерных (оба сибса больны), так и простых (один сибс болен, а один здоров) семей. При этом оцениваются шансы (вероятности) за и против сцепления в данной семье. Количественным показателем сцепления является логарифм соотношения шансов (правдоподобия) за и против сцепления – лод–балл (от английского «logarithm of odds ratio»). Сцепление считается подтвержденным, если лод–балл превысит значение 3.0 (т.е. вероятность совместного наследования признака и маркера в 1000 раз больше, нежели вероятность их раздельного наследования). Если ни на одной из проанализированных семей не обнаруживается статистически значимое сцепление, лод–балл рассчитывают, используя суммарные данные для нескольких семей. При этом необходим тщательный отбор семей из общей группы по фенотипическим признакам, позволяющих объединить их в одну подгруппу [1]. Также активно используют мультилокусный анализ сцепления, при котором рассчитывается соотношение вероятностей для каждого интервала между двумя соседними из десятков и сотен полиморфных маркеров, распределенных по всему геному. Интервалы, для которых значения лод–балла превышают 3.0, считают наиболее вероятными областями локализации гена, ассоциированного с конкретной патологией [1].
Наряду с анализом сцепления, при использовании которого необходимы полные семьи с двумя сибсами, в методе неравновесной передачи аллелей (TDT, transmission disequilibrium test) могут использоваться семьи с единственным потомком. Этот метод основан на анализе частот передачи аллелей полиморфных маркеров, расположенных в интересующей исследователя области генома, от гетерозиготных по данному маркеру родителей к больному потомку. Для сравнения используют аналогичные гетерозиготные семьи, но со здоровым потомком [2].
К сожалению, несмотря на преобладание в литературе в течение ряда лет работ, использующих данный метод, для многих заболеваний его результаты были редко воспроизводимы в независимых исследованиях [3]. Например, для сахарного диабета типа 2 из 40 обнаруженных локусов сцепления крайне малое число было подтверждено другими исследователями [4, 5]. Точность локализации локуса сцепления не превышает нескольких млн.п.н. и, как правило, требуются работы по более точному картированию с использованием однонуклеотидных полиморфизмов и блоков неравновесия по сцеплению [6, 7]. В целом, к идентификации этиологических вариантов заболеваний привели не так много работ (например, [8, 9]), как ожидалось, так как сигналы сцепления являются отражением эффектов редких генетических вариантов, в то время как анализ блоков неравновесия по сцеплению использует полиморфные маркеры с частотой редкого аллеля не менее 5%. Более того, из-за относительно малого количества семей и используемых микросателлитных маркеров большинство этих исследований оказалось неспособно обнаружить ассоциации, которые были найдены в более поздних работах с использованием методов «случай-контроль» и/или подхода «ген-кандидат».
Подход «ген-кандидат» позволяет сфокусироваться на одном или нескольких вариантах в области гена, продукт которого вероятно вовлечен в развитие патологии. Обычно исследование проводится на группах из нескольких сотен больных и здоровых индивидах или семьях. В первом случае проводят анализ распределения аллелей и генотипов исследуемого генетического маркера в выборке из неродственных здоровых лиц (популяционный контроль) и в группе больных (группа «случай») с тем, чтобы выявить значимые различия в частоте генетического маркера (аллеля). Генетический маркер считается ассоциированным с болезнью, если его частота среди больных значимо выше, нежели в контрольной выборке. Наличие ассоциации либо свидетельствует о прямой связи между исследованным локусом и наследственной патологией, либо в основе ассоциации может лежать неравновесное сцепление между маркерным локусом и локусом, обуславливающим развитие болезни, если эти локусы расположены достаточно близко друг от друга [1].
Ассоциация может оказаться мнимой за счет негомогенности популяции, малочисленности выборок, некорректности критериев отбора при формировании выборок больных и здоровых индивидов или неправильных представлений об этиопатогенезе заболевания. Нежелательных эффектов, связанных с этнической неоднородностью популяции, можно избежать, проводя анализ ассоциации в гомогенных изолированных популяциях. Группы сравнения должны быть правильно подобраны и хорошо охарактеризованы клинически и биохимически. Необходимо, чтобы они были гомогенны по негенетическим факторам риска [1].
Успехи в изучении распределения блоков неравновесия по сцеплению в геноме человека (проект HapMap, [10]) позволили поднять исследования ассоциаций на новый уровень – стало возможно выбирать максимально информативные наборы полиморфных маркеров (tag-SNP, рис. 1), характеризующие большинство наиболее распространенных полиморфных вариантов в интересующем регионе генома [11-15].
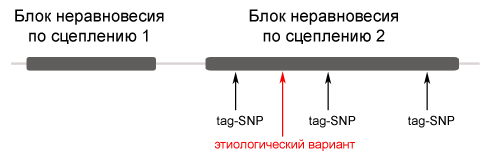
| Рис. 1. Схема выбора маркерных однонуклеотидных полиморфизмов (tag-SNP), позволяющих выявить область генома, содержащего полиморфный маркер, ассоциированный с изучаемым признаком. |
Развитие методов высокопроизводительного генотипирования привело к использованию сотен и тысяч полиморфных маркеров в рамках одного исследования. В настоящее время применяются биологические микрочипы высокой плотности, содержащие от 300 тыс. до 1 млн. полиморфных маркеров, равномерно распределенных по всему геному (табл. 1).
|
Таблица 1
|
||
|
Коммерчески доступные платформы генотипирования на микрочипах высокой плотности и охват полиморфных маркеров с частотой минорного аллеля более 5% в европейской популяции [16, 17].
|
||
| Платформа | Количество полиморфных маркеров | Охват, % |
| Illumina Human-1 |
более 109 000
|
26
|
| Illumina HumanHap300 |
317 511
|
75
|
| Affymetrix SNP Array 5.0 |
500 568
|
65
|
| Illumina HumanHap550 |
555 352
|
87
|
| Illumina Human610 |
620 901
|
89
|
| Illumina HumanHap650Y |
660 917
|
87
|
| Affymetrix SNP Array 6.0 |
более 800 000
|
83
|
| Illumina Human1M |
1 199 187
|
93
|
Благодаря большим размерам выборок данный подход (GWA, genome-wide association) обладает высокой статистической мощностью, воспроизводимостью и надежностью результатов (табл. 2). Но, несмотря на то, что он позволяет обнаруживать сравнительно слабые ассоциации с OR не менее 1.1-1.2, мета-анализы первой волны GWA-исследований показали, что для многих заболеваний удается надежно идентифицировать достаточно небольшую часть полиморфных маркеров, ассоциированных с патологией. Выяснилось, что генетическая предрасположенность к многофакторным заболеваниям не всегда объясняется распространенными полиморфизмами и для поиска этиологических вариантов (возможно, редких полиморфизмов или нуклеотидных повторов) потребуется дальнейшее исследование и секвенирование участков генома, выявленных в ходе GWA-исследований.
|
Таблица 2
|
||
|
Сравнение подходов «ген-кандидат» (GGAS, candidate gene association study) и полногеномного поиска (GWAS, genome-wide association study) в ассоциативных исследованиях [18].
|
||
| Свойство | GGAS | GWAS |
| Независимость от информации о функциях гена |
–
|
+
|
| Способность к коррекции популяционной гетерогенности |
–
|
+
|
| Максимальный охват в целом по геному |
–
|
+
|
| Максимальный охват области гена-кандидата |
+
|
–
|
|
Воспроизводимость |
+/–
|
+
|
|
Минимизация ложноположительных результатов |
–
|
+
|
| Минимизация ложноотрицательных результатов |
+
|
–
|
| Экономичность |
+
|
–
|
| Идентификация редких вариантов (частота минорного аллеля менее 5%) |
+
|
–
|
Результаты исследований «случай-контроль» позволяют оценить риск развития заболевания у конкретного индивида, но известно, что вклад различных генов в формирование генетической предрасположенности к патологиям существенно различается в разных популяциях, в то время как вычисление генетического риска для пациента требует наличия информации о распределении частот аллелей и генотипов для популяции, которой он принадлежит, что затрудняет использование данных, полученных для других популяций. Таким образом, необходимость проведения аналогичных исследований для каждой крупной популяции не вызывает сомнений.
1. Иванов В.И. 2005. Геномика - медицине [под ред. В. И. Иванова и Л. Л. Киселева]. М.: Академкнига; , 392 с.
2. Thomson G. 1995. Analysis of complex human genetic traits: an ordered-notation method and new tests for mode of inheritance. Am J Hum Genet. 57 (2), 474-86.
3. Panoutsopoulou K., Zeggini E. 2009. Finding common susceptibility variants for complex disease: past, present and future. Brief Funct Genomic Proteo-mic. , elp020.
4. McCarthy M.I. 2003. Growing evidence for diabetes susceptibility genes from genome scan data. Curr. Diab. Rep.3 (2), 159-167.
5. Guan W., Pluzhnikov A., Cox N.J., Boehnke M. 2008. Meta-analysis of 23 type 2 diabetes linkage studies from the International Type 2 Diabetes Linkage Analysis Consortium. Hum. Hered. 66 (1), 35-49.
6. Evans D.M., Cardon L.R. 2004. Guidelines for genotyping in genomewide linkage studies: single-nucleotide-polymorphism maps versus microsatellite maps. Am. J. Hum. Genet. 75 (4), 687-692.
7. John S., Shephard N., Liu G. et al. 2004. Whole-genome scan, in a complex disease, using 11,245 single-nucleotide polymorphisms: comparison with microsatellites. Am. J. Hum. Genet. 75 (1), 54-64.
8. Ogura Y., Bonen D.K., Inohara N. et al. 2001. A frameshift mutation in NOD2 associated with susceptibility to Crohn's disease. Nature. 411 (6837), 603-606.
9. Hugot J.P., Chamaillard M., Zouali H. et al. 2001. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn's disease. Nature. 411 (6837), 599-603.
10. Frazer K.A., Ballinger D.G., Cox D.R. et al. 2007. A second generation hu-man haplotype map of over 3.1 million SNPs. Nature. 449 (7164), 851-861.
11. Ke X., Miretti M.M., Broxholme J., et al. 2005. A comparison of tagging methods and their tagging space. Hum. Mol. Genet. 14 (18), 2757-2767.
12. Johnson G.C., Esposito L., Barratt B.J. et al. 2001. Haplotype tagging for the identification of common disease genes. Nat Genet. 29 (2), 233-7.
13. Gabriel S.B., Schaffner S.F., Nguyen H. et al. 2002. The Structure of Haplotype Blocks in the Human Genome.Science. 296 (5576), 2225-2229.
14. Carlson C.S., Eberle M.A., Rieder M.J. et al. 2004. Selecting a maximally informative set of single-nucleotide polymorphisms for association analyses using linkage disequilibrium. Am. J. Hum. Genet. 74 (1), 106-120.
15. Halldorsson B.V., Istrail S., De La Vega F.M. 2004. Optimal selection of SNP markers for disease association studies. Hum Hered. 58 (3-4), 190-202.
16. Barrett J.C., Cardon L.R. 2006. Evaluating coverage of genome-wide association studies. Nat. Genet. 38 (6), 659-662.
17. Li M., Li C., Guan W. 2008. Evaluation of coverage variation of SNP chips for genome-wide association studies.Eur. J. Hum. Genet. 16 (5), 635-643.
18. Sethupathy P., Collins F.S. 2008. MicroRNA target site polymorphisms and human disease. Trends Genet. 24 (10), 489-497.
Источник: Ген Эксперт
Вернуться назад
 Разработка сайта
Разработка сайта